
{kind=link}
AlphaFold is a Google DeepMind AI system that predicts protein structure. Similar to proteins, we need to know the structure of a material to predict its properties. What we need is an AlphaFold for materials.
An AI program created by nanoengineers at the University of California San Diego’s Jacobs School of Engineering can nearly instantly identify the structure and dynamic characteristics of any material, whether it is new or existing.
The system, known as M3GNet, was used to create matterverse.ai, a library of more than 31 million materials that have not yet been synthesized but whose characteristics have been predicted by machine learning algorithms. Matterverse.ai makes it easier to find new technology materials with extraordinary features.
For the purpose of finding safer and more energy-dense electrodes and electrolytes for rechargeable lithium-ion batteries, the M3GNet team, led by UC San Diego nanoengineering professor Shyue Ping Ong, combines matterverse.ai and the new capabilities of M3GNet. The work has been detailed in a study published in Nature Computational Science today.
The arrangement of atoms in a substance determines its qualities. However, the methods now in use to achieve such arrangements are either expensive or inefficient for several reasons.
“Similar to proteins, we need to know the structure of a material to predict its properties.” adds Ong. “What we need is an AlphaFold for materials.”
AlphaFold is a Google DeepMind AI system that predicts protein structure. Ong and his colleagues coupled graph neural networks with many-body interactions to create a deep learning architecture that operates uniformly and accurately across all of the elements in the periodic table to create the equivalent for materials.
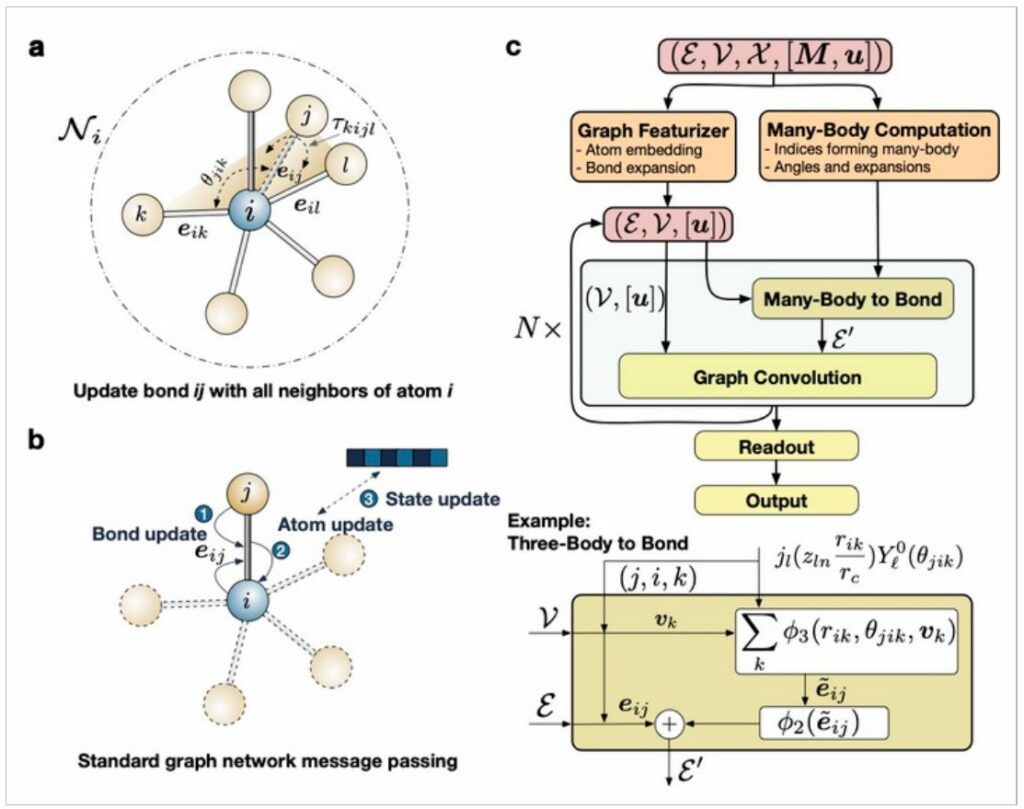
“Mathematical graphs are really natural representations of a collection of atoms,” adds first author Chi Chen. “Using graphs, we can represent the full complexity of materials without being subject to the combinatorial explosion of terms in traditional formalisms.”
The researchers utilized the massive database of materials energy, pressures, and stresses gathered in the Materials Project over the last decade to train their model. The outcome is the M3GNet interatomic potential (IAP), which can calculate the energies and forces of any atom collection. Matterverse.ai was created by performing combinatorial elemental substitutions on over 5,000 structural prototypes from the Inorganic Crystal Structure Database (ICSD). For property prediction, the M3GNet IAP was then employed to create the equilibrium crystal structure, a process known as “relaxation.”
Over a million matterverse.ai’s 31 million materials are theoretically stable. Using a multi-fidelity method they previously created, Ong and his colleagues want to significantly increase not just the amount of materials but also the number of ML-predicted characteristics, including high-value qualities with modest data volumes.
The M3GNet IAP has several uses in dynamic simulations of materials and property predictions in addition to structural relaxations.
“For instance, we are often interested in how fast lithium ions diffuse in a lithium-ion battery electrode or electrolyte. The faster the diffusion, the more quickly you can charge or discharge a battery,” Ong adds. “We have shown that the M3GNet IAP can be used to predict the lithium conductivity of a material with good accuracy. We truly believe that the M3GNet architecture is a transformative tool that can greatly expand our ability to explore new material chemistries and structures.”
The M3GNet team has made the framework available on Github as open-source Python code in order to encourage more people to utilize M3GNet. Since publishing the preprint on Arxiv in February 2022, the team has garnered attention from both academia and industrial researchers. The M3GNet IAP will be included as a tool in commercial materials modeling software, according to plans.